Variations
What Are Variations?
Variations refer to post-approval regulatory changes made to an already authorized medicinal product. After a product receives marketing authorization, any modification related to quality, safety, efficacy, labeling, manufacturing, packaging, or administrative details must be formally submitted to health authorities for approval or notification.
In the European regulatory framework governed by the European Medicines Agency and national competent authorities, variations are classified based on the potential impact of the change on product quality, safety, or efficacy.
Concept of Variations
A marketing authorization is granted based on specific approved conditions. These include formulation, manufacturing site, specifications, labeling text, indication, and dosage. Any change to these approved conditions requires regulatory assessment to ensure the benefit–risk balance remains favorable.
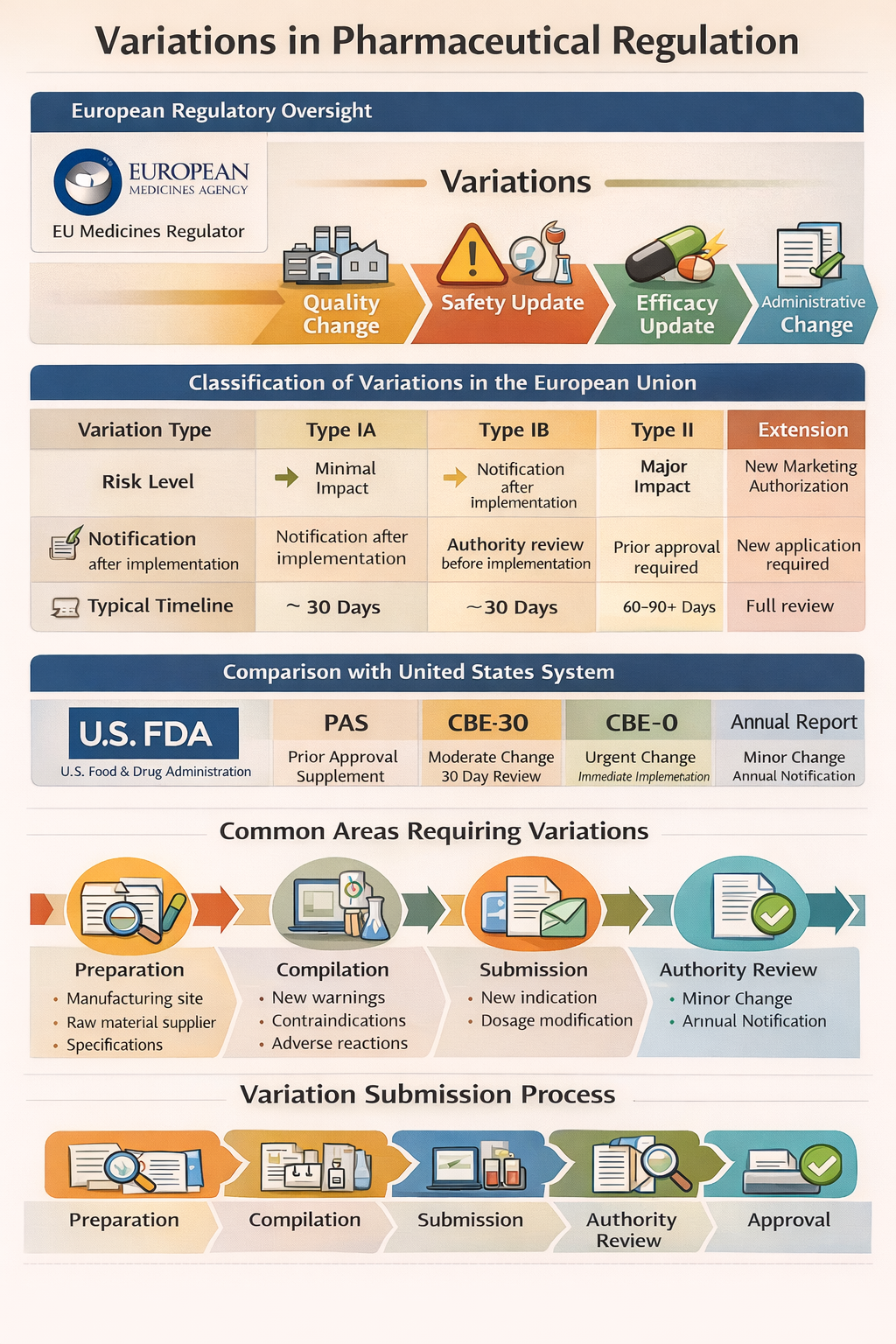
Classification of Variations in the European Union
| Variation Type | Risk Level | Approval Requirement | Typical Timeline |
|---|---|---|---|
| Type IA | Minimal impact | Notification after implementation | 30 days |
| Type IA IN | Immediate notification | Implement first, notify immediately | 30 days validation |
| Type IB | Minor impact | Authority review before implementation | ~30 days |
| Type II | Major impact | Prior approval required | 60–90+ days |
| Extension | Significant change (new MA) | New application required | Full review |
Type IA Variation
This category includes administrative or minor quality changes that do not significantly impact product quality or safety. Examples include change in company address, tightening of specifications, or minor manufacturing process adjustments. These can generally be implemented before notifying the authority.
Type IB Variation
These are minor but not negligible changes. If the authority does not object within the review period, the change may proceed. Examples include certain manufacturing site changes or updates to test methods.
Type II Variation
These involve significant changes that may affect quality, safety, or efficacy. Examples include addition of a new indication, major safety labeling update, new strength addition, or significant manufacturing process change. These require detailed assessment and formal approval before implementation.
Extension Application
An extension is treated almost like a new marketing authorization. Examples include new pharmaceutical form, new route of administration, or new strength requiring separate evaluation.
Comparison with United States System
In the United States, under the U.S. Food and Drug Administration, post-approval changes are categorized differently:
| Category | Description |
|---|---|
| Prior Approval Supplement (PAS) | Major changes requiring approval before implementation |
| CBE-30 | Changes Being Effected in 30 days |
| CBE-0 | Immediate implementation with notification |
| Annual Report | Minor changes reported annually |
While terminology differs, the risk-based principle is similar: higher risk changes require prior approval.
Common Areas Requiring Variations
Quality-related changes such as manufacturing site transfer, equipment upgrade, raw material supplier change, or specification revision.
Safety-related changes including addition of warnings, contraindications, or adverse reactions based on pharmacovigilance data.
Efficacy-related changes such as new indication or dosing modification.
Administrative updates including MAH name change or artwork modification.
Variation Dossier Structure
A variation submission typically includes:
Updated application form
Revised labeling (if applicable)
Supporting scientific justification
Comparative data (if quality change)
Risk assessment
Updated CTD modules
Impact on Labeling Lifecycle
Many variations trigger label updates. For example, a Type II safety variation may require SmPC, PIL, and packaging text changes across multiple markets. Therefore, variation management and label lifecycle management are closely linked.
Regulatory Strategy Considerations
Companies must determine the correct variation classification before submission. Incorrect classification can lead to rejection or delay. Global companies often coordinate updates centrally through the Company Core Data Sheet to maintain consistency across markets.
Inspection and Compliance Risk
Failure to submit a required variation, late submission, or implementation without approval can result in regulatory findings, warning letters, or product recall.
Variations are therefore a structured regulatory mechanism ensuring that any post-approval change maintains product quality, safety, and efficacy while preserving regulatory compliance throughout the product lifecycle.
