Clinical Trial Phases (Phase I, II, III, IV)
Clinical Trial Phases (Phase I, II, III, IV)
Introduction
Clinical trials are structured research studies conducted in human participants to evaluate the safety, efficacy, quality, and overall benefit–risk profile of investigational medicinal products. They represent a critical transition from laboratory and animal research to human therapeutic application. Clinical development is conducted in sequential phases, each designed with specific objectives, participant populations, and regulatory expectations.
Clinical trials must comply with ethical and scientific standards established under Good Clinical Practice. Regulatory oversight is provided by authorities such as the U.S. Food and Drug Administration and the Central Drugs Standard Control Organization. Approval to conduct clinical trials requires prior authorization based on robust pre-clinical data, and all trial activities must adhere to approved protocols and ethical review processes.
The development process is divided into four major phases: Phase I, Phase II, Phase III, and Phase IV. Each phase builds upon data generated in the preceding stage.
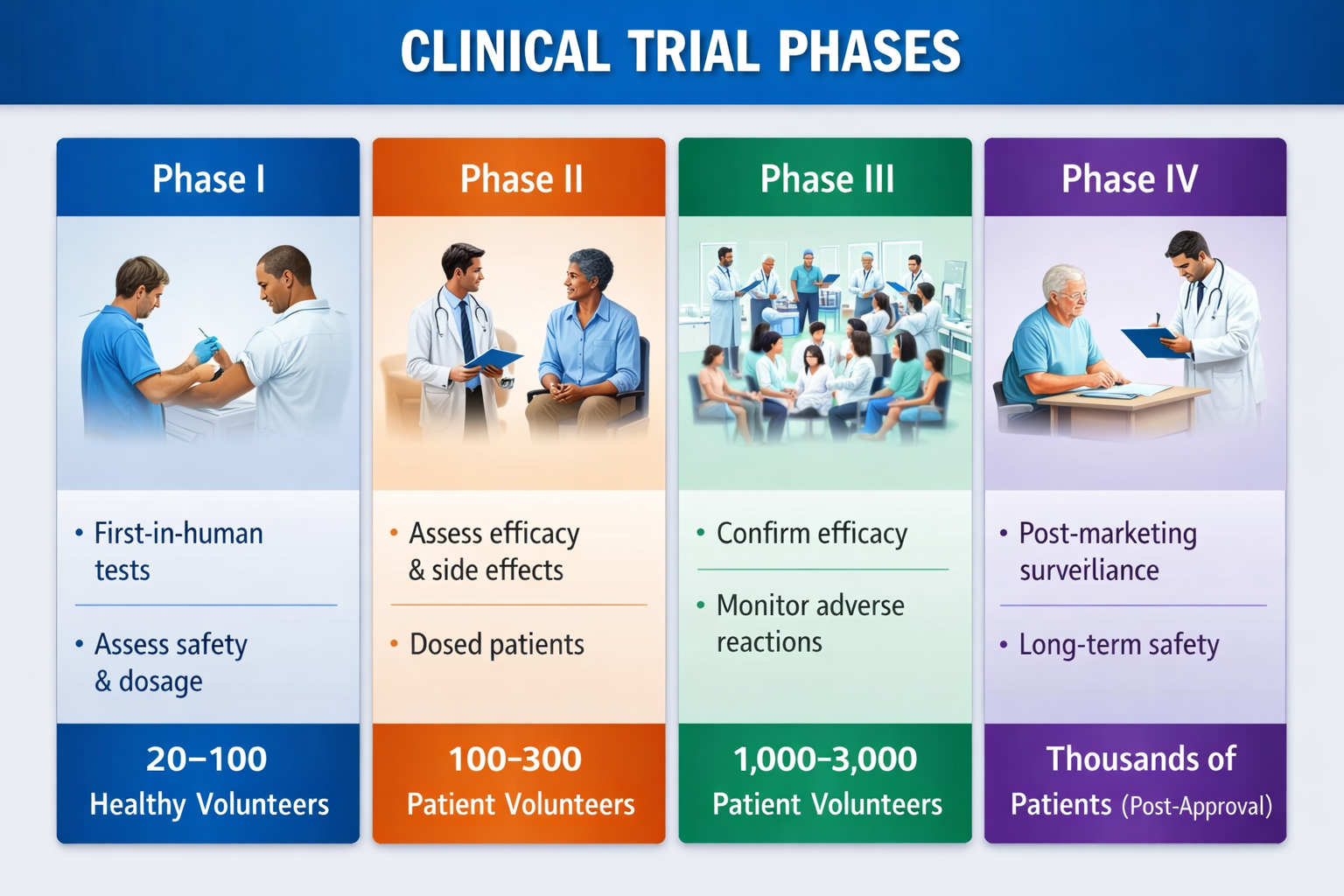 Overview of Clinical Trial Phases
Overview of Clinical Trial Phases
The following table summarizes the key structural differences between each clinical trial phase:
| Phase | Participant Population | Primary Objective | Approximate Sample Size | Regulatory Significance |
|---|---|---|---|---|
| Phase I | Healthy volunteers or selected patients | Evaluate safety and dosage | 20–100 | Establish safe dosing range |
| Phase II | Patients with target disease | Assess preliminary efficacy and safety | 100–300 | Optimize therapeutic dose |
| Phase III | Large patient population | Confirm efficacy and monitor adverse reactions | 1000–3000+ | Basis for marketing approval |
| Phase IV | Approved drug users | Monitor long-term safety and real-world effectiveness | Variable | Post-marketing surveillance |
Each phase is carefully designed to minimize patient risk while maximizing scientific reliability.
Phase I – First-in-Human Studies
Phase I represents the first time the investigational drug is administered to humans. These studies are generally conducted in a small number of healthy volunteers, although patients may be included in cases involving oncology or life-threatening conditions.
The primary objective of Phase I is safety assessment. Researchers evaluate tolerability, adverse effects, and pharmacokinetic parameters including absorption, distribution, metabolism, and excretion. Pharmacodynamic responses are also observed to understand how the drug interacts with biological systems.
Dose escalation designs are commonly used in this phase. Single Ascending Dose and Multiple Ascending Dose study formats allow researchers to gradually increase dosage while monitoring safety. Determination of the maximum tolerated dose and identification of dose-limiting toxicities are critical outcomes. The data generated establish the recommended dose for Phase II studies.
Phase II – Therapeutic Exploratory Studies
Phase II trials involve a larger group of participants diagnosed with the target disease. The focus shifts from pure safety to evaluating preliminary efficacy while continuing safety monitoring.
These trials are often randomized and controlled. They aim to determine whether the drug produces the intended therapeutic effect under defined clinical conditions. Dose optimization is a key objective, requiring researchers to evaluate dose frequency, treatment duration, and therapeutic response.
Phase II studies are often subdivided into Phase IIa and Phase IIb. Phase IIa primarily focuses on dose-finding and early signs of efficacy, while Phase IIb confirms therapeutic activity at selected doses. Identification of common adverse drug reactions also occurs during this stage.
Successful completion of Phase II indicates that the drug demonstrates sufficient therapeutic promise to justify larger confirmatory trials.
Phase III – Therapeutic Confirmatory Studies
Phase III trials are large-scale, multicenter studies conducted across multiple geographical locations. These trials involve a significantly larger patient population and are designed to confirm efficacy and safety in diverse clinical settings.
In Phase III, the investigational drug is typically compared with either an established standard therapy or a placebo. Randomized controlled designs are frequently used to eliminate bias and ensure statistical reliability.
Extensive safety data are collected to identify less common adverse effects that may not have appeared in earlier phases. Special population groups, such as elderly patients, pediatric patients, and individuals with comorbidities, may also be studied.
The data generated from Phase III trials form the scientific foundation for submission of a New Drug Application or Marketing Authorization Application. Regulatory authorities use these results to assess the overall benefit–risk profile of the drug and determine whether approval is justified.
Phase IV – Post-Marketing Surveillance
Phase IV trials occur after regulatory approval and market introduction of the drug. Although the product has been authorized for use, long-term safety and real-world performance must continue to be monitored.
Post-marketing studies focus on identifying rare or delayed adverse drug reactions that may not have been detected in earlier clinical phases due to limited sample sizes. Real-world effectiveness data are also evaluated to assess outcomes under routine medical practice.
Phase IV may also involve studies exploring new therapeutic indications, additional dosage forms, or expanded patient populations. Pharmacovigilance programs and risk management plans are implemented to ensure continuous monitoring of safety signals.
Ethical and Regulatory Framework
Clinical trials must operate within a strict ethical framework. Participants are required to provide informed consent after being fully informed about potential risks and benefits. Study protocols must be reviewed and approved by institutional ethics committees before initiation.
All trial activities must align with Good Clinical Practice guidelines, ensuring data credibility and participant safety. Regulatory authorities conduct inspections to verify compliance with clinical, documentation, and reporting standards.
The following table summarizes essential regulatory characteristics of clinical trials:
| Regulatory Element | Description |
|---|---|
| Good Clinical Practice | International ethical and scientific quality standard |
| Informed Consent | Participant agreement after risk disclosure |
| Protocol Adherence | Strict compliance with approved study design |
| Safety Reporting | Timely reporting of adverse events |
| Regulatory Oversight | Inspections and approvals by national authorities |
