Innovator vs Generic Approval
Overview
The approval process for innovator drugs and generic drugs differs significantly in terms of development requirements, regulatory data, timelines, cost, and scientific evaluation. Regulatory authorities such as the U.S. Food and Drug Administration, the European Medicines Agency, the Central Drugs Standard Control Organization, Health Canada, and the Therapeutic Goods Administration follow distinct regulatory pathways for these two categories.
Innovator drugs introduce new active substances and require full scientific development, while generic drugs are approved based on equivalence to an already approved reference product.
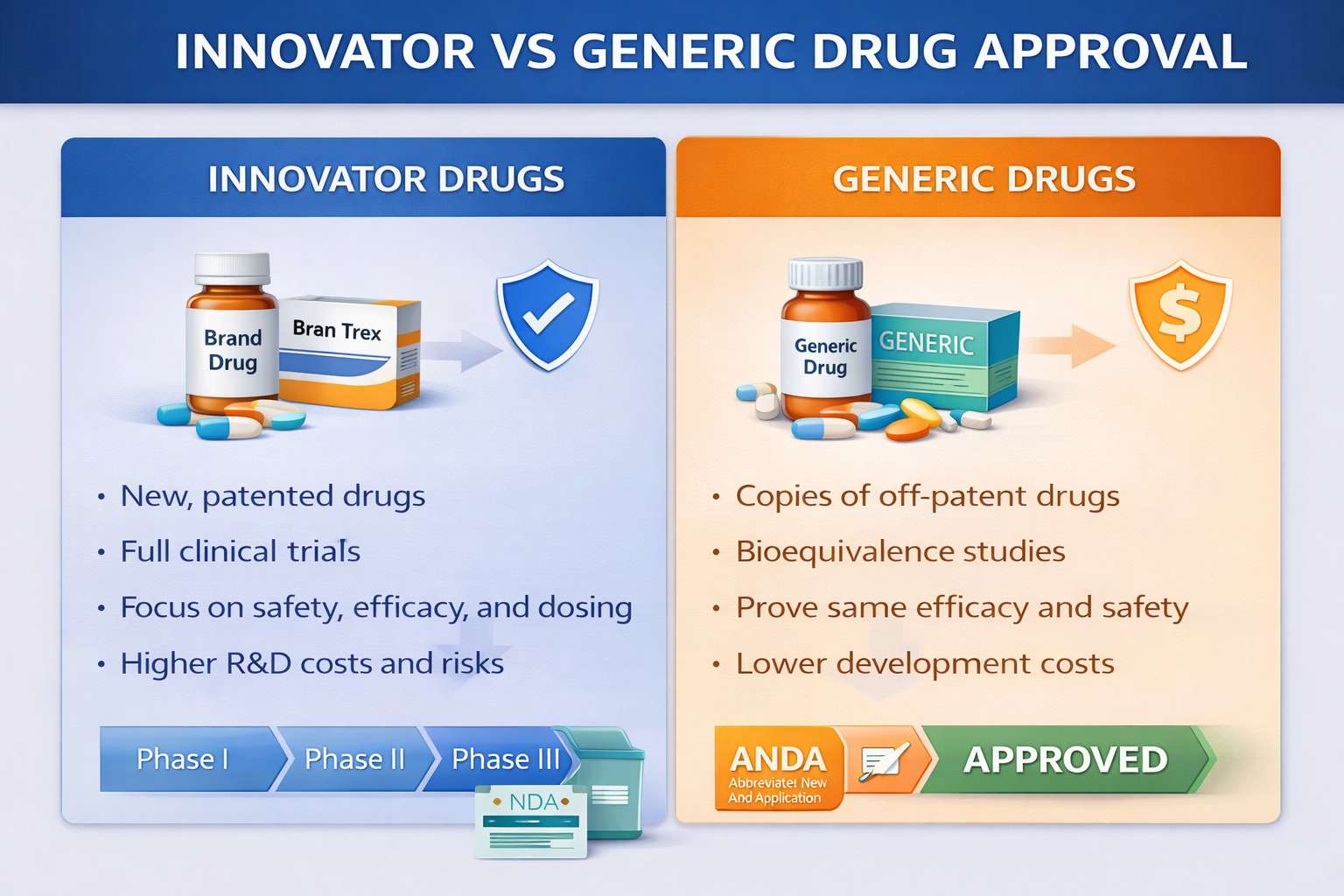
Innovator Drug Approval
An innovator drug, also known as an originator product, contains a new active pharmaceutical ingredient that has not been previously approved. These drugs require complete research and development and are usually protected by patents and regulatory exclusivity. Because of the extensive scientific studies involved, innovator drugs require significant investment and longer development timelines.
The development process begins with preclinical studies, which include pharmacology and toxicology testing in laboratory and animal models. These studies are followed by a full clinical development program. Phase I trials evaluate safety and dosage in healthy volunteers. Phase II trials assess efficacy and optimize dosing in patients. Phase III trials confirm safety and effectiveness in large patient populations.
In addition to clinical data, companies must submit complete Chemistry, Manufacturing, and Controls information, stability studies, a risk management plan, and proposed labeling and prescribing information.
Different regulatory agencies use specific application types for innovator drugs. In the United States, the application is called a New Drug Application. In the European Union and the United Kingdom, it is submitted as a Marketing Authorization Application. In Canada, it is known as a New Drug Submission. In India, the application is submitted to CDSCO as a new drug approval request.
During evaluation, regulators focus on proof of safety and efficacy, a comprehensive benefit–risk assessment, manufacturing quality, compliance with Good Manufacturing Practices, and post-marketing safety commitments.
Generic Drug Approval
A generic drug is a pharmaceutical product that is equivalent to an already approved innovator drug whose patent or exclusivity period has expired. Generic drugs contain the same active ingredient, dosage form, strength, and route of administration as the reference product and must demonstrate therapeutic equivalence.
The development process for generics is shorter and less expensive because the safety and efficacy of the active ingredient have already been established. Instead of full clinical trials, the sponsor must conduct a bioequivalence study to show that the generic product delivers the same amount of active ingredient into the bloodstream at the same rate as the reference drug.
The submission includes bioequivalence data, pharmaceutical equivalence information, complete manufacturing and quality details, and stability studies. Full Phase I, II, and III clinical trials are generally not required.
Regulatory application types for generics differ by region. In the United States, the submission is called an Abbreviated New Drug Application. In the European Union, it is submitted as a generic Marketing Authorization Application. In Canada, it is called an Abbreviated New Drug Submission. In India, the generic application is submitted to CDSCO.
During evaluation, regulators focus on demonstrating bioequivalence, ensuring pharmaceutical equivalence, verifying manufacturing and quality compliance, and confirming that labeling is consistent with the reference product.
Key Differences Between Innovator and Generic Approvals
The development timeline for innovator drugs is significantly longer, typically ranging from ten to fifteen years. This extended period is due to the need for extensive research, clinical trials, and regulatory evaluations. In contrast, generic drug development usually takes two to five years because it relies on existing scientific knowledge about the reference product.
The cost of developing an innovator drug is very high because it involves discovery research, multiple clinical trial phases, and extensive regulatory documentation. Generic drugs are much less expensive to develop because they do not require full clinical efficacy trials.
Innovator drugs require a complete clinical development program, including Phase I, II, and III trials. Generic drugs generally require only bioequivalence studies.
Patent protection is another major difference. Innovator drugs are protected by patents and regulatory exclusivity for a defined period. Generic drugs are approved only after these protections expire.
Scientific risk is also different. Innovator drug development carries high uncertainty and regulatory risk because the active substance is new. Generic drug approval involves lower risk because the reference product’s safety and efficacy are already established.
Overall, innovator and generic drug approvals follow different regulatory pathways designed to balance innovation with affordability. Innovator pathways encourage the development of new therapies, while generic pathways promote access to cost-effective medicines once patents expire.
