Clinical Protocol Submission
What is Clinical protocol submission?
Clinical protocol submission is one of the most critical regulatory activities under an active Investigational New Drug (IND) with the US Food and Drug Administration. A clinical protocol is not merely a scientific plan; it is a legally binding document that defines how human subjects will be exposed to an investigational product. From a Regulatory Affairs perspective, protocol submission is both a compliance obligation and a strategic development decision that directly influences regulatory review, clinical execution, and eventual marketing approval.
Regulatory Basis and Timing
Under 21 CFR 312.30, a new protocol must be submitted to the FDA as a Protocol Amendment before implementation if the IND is already in effect. If the protocol is included in the original IND submission, it becomes part of the initial 30-day safety review window. No clinical trial may begin until either the 30-day review period has elapsed without a clinical hold or explicit authorization has been granted
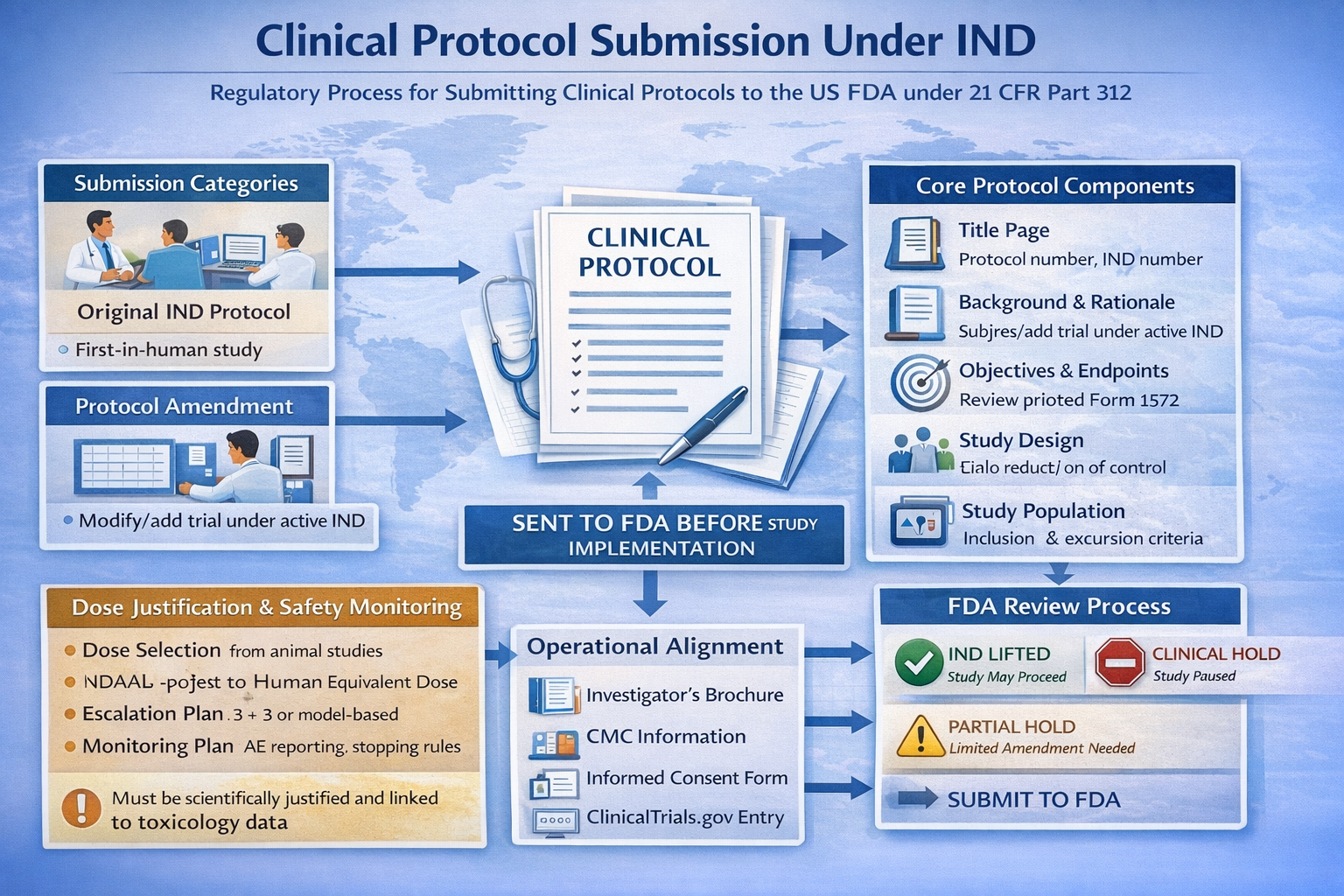
The regulatory classification of protocol submissions is structured as follows.
| Submission Category | When Used | FDA Review Implication |
|---|---|---|
| Original IND Protocol | First-in-human study | Subject to 30-day safety review |
| Protocol Amendment – New Study | Addition of new clinical trial | Review prior to implementation |
| Protocol Amendment – Change in Study | Modification to ongoing study | Requires FDA notification before change |
| Protocol Amendment – New Investigator | Addition of new clinical site | Must include updated Form 1572 |
Core Structure of a Clinical Protocol
A job-ready Regulatory Affairs professional must ensure that each protocol is scientifically sound, internally consistent, and aligned with nonclinical and CMC data already submitted in the IND. The protocol must clearly demonstrate subject protection, dose justification, and methodological rigor.
The structured components of a clinical protocol are summarized below.
| Protocol Section | Key Content | Regulatory Objective |
|---|---|---|
| Title Page | Study title, protocol number, IND number | Administrative identification |
| Background and Rationale | Scientific justification | Link to nonclinical findings |
| Objectives and Endpoints | Primary and secondary objectives | Define measurable outcomes |
| Study Design | Randomization, control, blinding | Ensure validity and bias control |
| Study Population | Inclusion and exclusion criteria | Protect vulnerable subjects |
| Dose and Administration | Dose levels, escalation plan | Align with toxicology data |
| Safety Monitoring | AE reporting, stopping rules | Risk mitigation |
| Statistical Analysis Plan | Sample size and methods | Ensure reliable interpretation |
| Data Handling and Recordkeeping | Source documentation and data capture | Maintain regulatory traceability |
Dose Justification and Risk Mitigation
Dose selection is one of the most scrutinized elements of a Phase I protocol. It must be derived from the No Observed Adverse Effect Level in animal studies and adjusted to Human Equivalent Dose using appropriate safety factors. The escalation strategy, whether traditional 3+3 design or model-based design, must be justified scientifically.
If toxicology data suggest organ-specific risk, the protocol must incorporate enhanced monitoring parameters. For example, hepatotoxicity findings require scheduled liver function testing, predefined stopping rules, and clear adverse event grading criteria.
Safety Reporting Integration
Clinical protocol submission must align with IND safety reporting requirements. Serious and unexpected suspected adverse reactions must be reported within 7 or 15 calendar days depending on severity. The protocol should define investigator responsibilities for immediate safety reporting and describe how data will be reviewed by safety committees or Data Monitoring Boards.
Operational Documentation Alignment
Regulatory professionals must ensure consistency across the following documents before submission.
| Document | Alignment Requirement |
|---|---|
| Investigator’s Brochure | Must reflect current safety data |
| CMC Information | Must match dosage form and strength |
| Informed Consent Form | Must reflect protocol risks |
| ClinicalTrials.gov Entry | Must mirror protocol design |
Discrepancies between these documents are a common cause of regulatory queries and inspection findings.
FDA Review Considerations
During the review process, the FDA evaluates whether the protocol adequately protects subjects and whether the scientific design supports meaningful data generation. The agency may place the study on clinical hold if it identifies unreasonable risk, inadequate monitoring, insufficient investigator qualifications, or flawed design.
Lifecycle Management
Once implemented, protocols may require amendments due to safety findings, dosing adjustments, or changes in study population. Each amendment must be clearly identified, version-controlled, and submitted before implementation unless the change is required to eliminate immediate hazard to subjects.
| Amendment Type | Example | Regulatory Action |
|---|---|---|
| Safety Amendment | Reduced dose after toxicity signal | Submit before implementation |
| Administrative Amendment | Correction of typographical error | Submit for documentation |
| Design Amendment | Addition of new cohort | Requires FDA review |
Job-Ready Competencies
A Regulatory Affairs Associate must develop the ability to critically review protocols for regulatory compliance, cross-check scientific consistency, coordinate with clinical development teams, and manage submission timelines. Familiarity with electronic submission standards and document version control systems is essential. Experience in preparing protocol amendments and responding to FDA information requests significantly enhances employability.
Conclusion
Clinical protocol submission under an IND is a highly regulated, scientifically rigorous process that directly impacts patient safety and development timelines. It requires alignment of nonclinical evidence, manufacturing quality, clinical methodology, and ethical oversight. Mastery of protocol submission strategy and lifecycle management is a foundational skill for professionals pursuing careers in global Drug Regulatory Affairs.
