IND Amendments
What are IND Amendments?
IND Amendments are formal submissions made to an active Investigational New Drug application to notify the FDA of changes or to provide new information during the clinical development lifecycle. Under 21 CFR 312.30 and 312.31, sponsors are legally obligated to maintain an updated IND. From a regulatory affairs perspective, amendments are not administrative formalities; they are controlled risk-management communications that ensure subject safety, product quality, and regulatory transparency throughout clinical development.
An IND is a living regulatory file. Once the original IND becomes effective, development rarely proceeds without modifications. New protocols are added, safety findings emerge, manufacturing processes evolve, and additional data become available. Each such change must be appropriately categorized and submitted in the correct amendment type to avoid compliance risk.
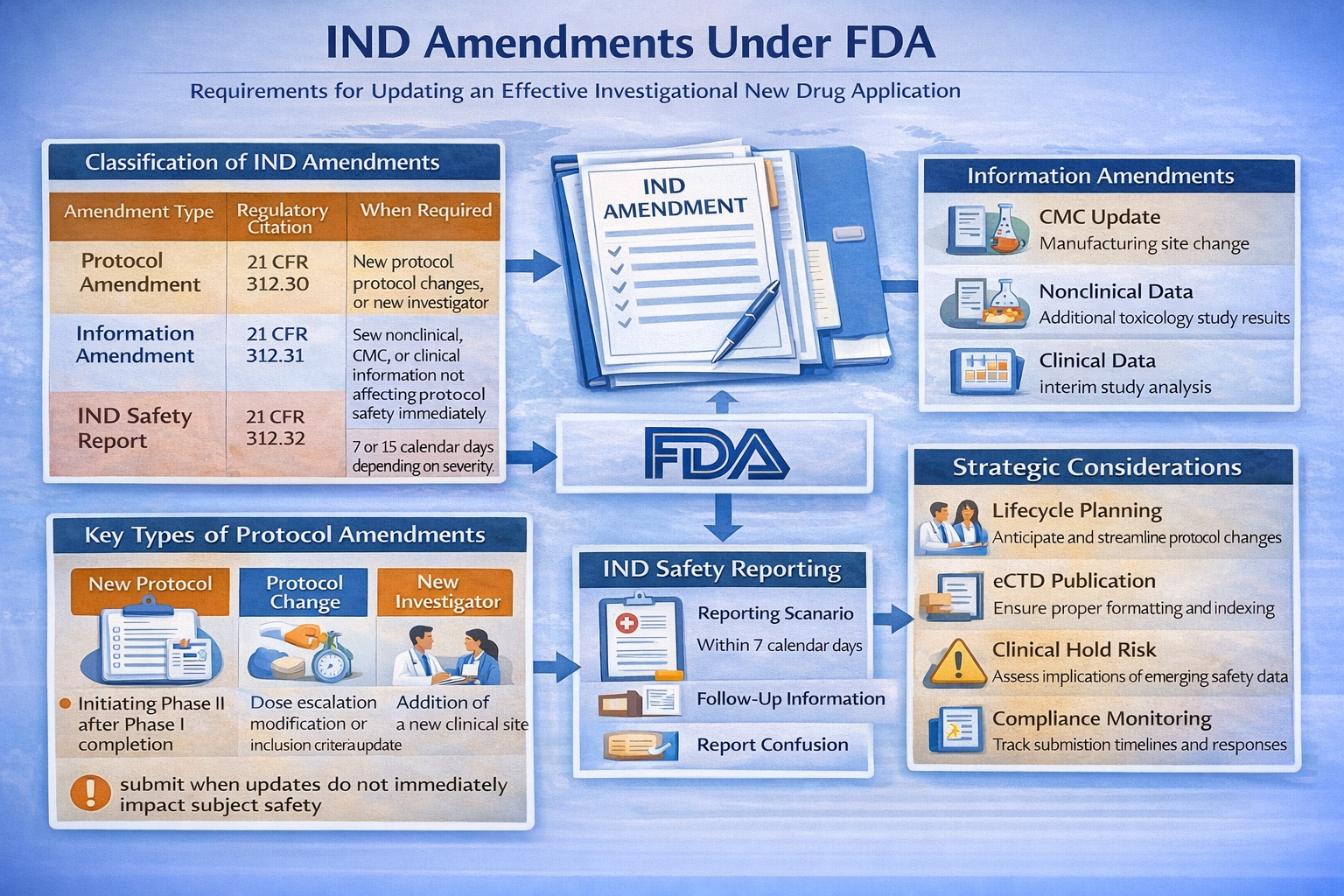
Classification of IND Amendments
| Amendment Type | Regulatory Citation | When Required | FDA Review Expectation |
|---|---|---|---|
| Protocol Amendment | 21 CFR 312.30 | New protocol, protocol changes, or new investigator | Must be submitted before implementation (except safety-driven changes) |
| Information Amendment | 21 CFR 312.31 | New nonclinical, CMC, or clinical information not affecting protocol safety immediately | Submitted as needed; no 30-day waiting period |
| IND Safety Report | 21 CFR 312.32 | Serious and unexpected suspected adverse reactions | 7 or 15 calendar days depending on severity |
Protocol Amendments
Protocol Amendments are the most common type encountered by Regulatory Affairs Associates. These are required when a sponsor introduces a new clinical study under the same IND, modifies an existing protocol, or adds a new investigator to an ongoing study.
The three categories of protocol amendments are summarized below.
| Subtype | Practical Example | Submission Timing |
|---|---|---|
| New Protocol | Initiating Phase II after Phase I completion | Before study initiation |
| Protocol Change | Dose escalation modification or inclusion criteria update | Before implementation unless immediate hazard |
| New Investigator | Addition of a new clinical site | Within 30 days of investigator participation |
A key compliance principle is that changes intended to eliminate immediate hazards to subjects may be implemented first and reported afterward. However, documentation must clearly justify the urgency. Regulatory professionals must coordinate closely with clinical operations to ensure no study activity proceeds without appropriate FDA notification.
Information Amendments
Information Amendments are used to submit additional supporting data that does not immediately impact subject safety or protocol conduct. These may include updated stability data, revised manufacturing specifications, additional pharmacology findings, or updated Investigator’s Brochure content.
| Category | Typical Content | Regulatory Risk Level |
|---|---|---|
| CMC Update | Manufacturing site change, analytical method refinement | Moderate to high |
| Nonclinical Data | Additional toxicology study results | High if safety relevant |
| Clinical Data | Interim study analysis | Moderate |
Regulatory professionals must exercise judgment in determining whether new data require a protocol amendment instead of an information amendment. Misclassification can result in regulatory findings during inspections.
IND Safety Reports
IND Safety Reporting represents the most time-sensitive amendment category. Sponsors must report serious and unexpected suspected adverse reactions associated with the investigational product.
| Reporting Scenario | Timeline |
|---|---|
| Fatal or life-threatening unexpected adverse reaction | Within 7 calendar days |
| Other serious and unexpected suspected adverse reaction | Within 15 calendar days |
| Follow-up information | As soon as available |
A well-functioning pharmacovigilance system is critical. Regulatory teams must ensure alignment between global safety databases and U.S. reporting obligations. Failure to report within mandated timelines can lead to inspection observations or enforcement actions.
Strategic Considerations in Amendment Management
In multinational pharmaceutical companies such as Pfizer, Novartis, and Roche, amendment planning is proactive rather than reactive. Lifecycle management strategy anticipates protocol evolution across development phases. For example, dose expansion cohorts are often pre-planned to minimize multiple sequential amendments. Regulatory Affairs must work within cross-functional development teams to streamline submissions while maintaining compliance.
Amendments must be submitted electronically in eCTD format through the FDA Electronic Submissions Gateway. Each amendment requires a cover letter clearly identifying the submission type, IND number, and purpose. Proper indexing within Module 1 ensures traceability during FDA review and inspection.
FDA Review Outcomes Following Amendments
While most amendments do not trigger a formal review clock like the original IND, the FDA retains authority to impose a clinical hold at any time if new information raises safety concerns.
| FDA Action | Implication |
|---|---|
| No Objection | Study continues as planned |
| Information Request | Sponsor must provide clarification |
| Clinical Hold | Study initiation or continuation suspended |
A regulatory professional must continuously assess whether emerging data could alter the risk-benefit profile of the investigational product.
Job-Ready Competencies for IND Amendment Handling
An entry-level Regulatory Affairs Associate should master amendment classification, regulatory citation knowledge, eCTD lifecycle publishing basics, cover letter drafting, and cross-functional coordination. Precision in documentation, timeline tracking, and regulatory intelligence monitoring is essential. Familiarity with FDA guidance documents and Good Clinical Practice principles ensures that amendments are scientifically justified and ethically compliant.
In conclusion, IND Amendments are central to lifecycle regulatory management. They reflect the dynamic nature of clinical development and demand structured regulatory oversight. Mastery of amendment processes distinguishes competent regulatory professionals and directly influences development timelines, compliance posture, and ultimately, successful drug approval pathways.
