Regulatory Approval Pathways
Overview
Regulatory approval pathways are structured processes established by health authorities to evaluate and authorize medicinal products before they are marketed. These pathways ensure that products meet required standards of safety, efficacy, and quality, while also allowing flexibility for innovative treatments or medicines needed in urgent public health situations.
Major regulatory agencies such as the U.S. Food and Drug Administration, European Medicines Agency, Central Drugs Standard Control Organization, Medicines and Healthcare products Regulatory Agency, Health Canada, and the Therapeutic Goods Administration offer different approval routes depending on product type and therapeutic need.
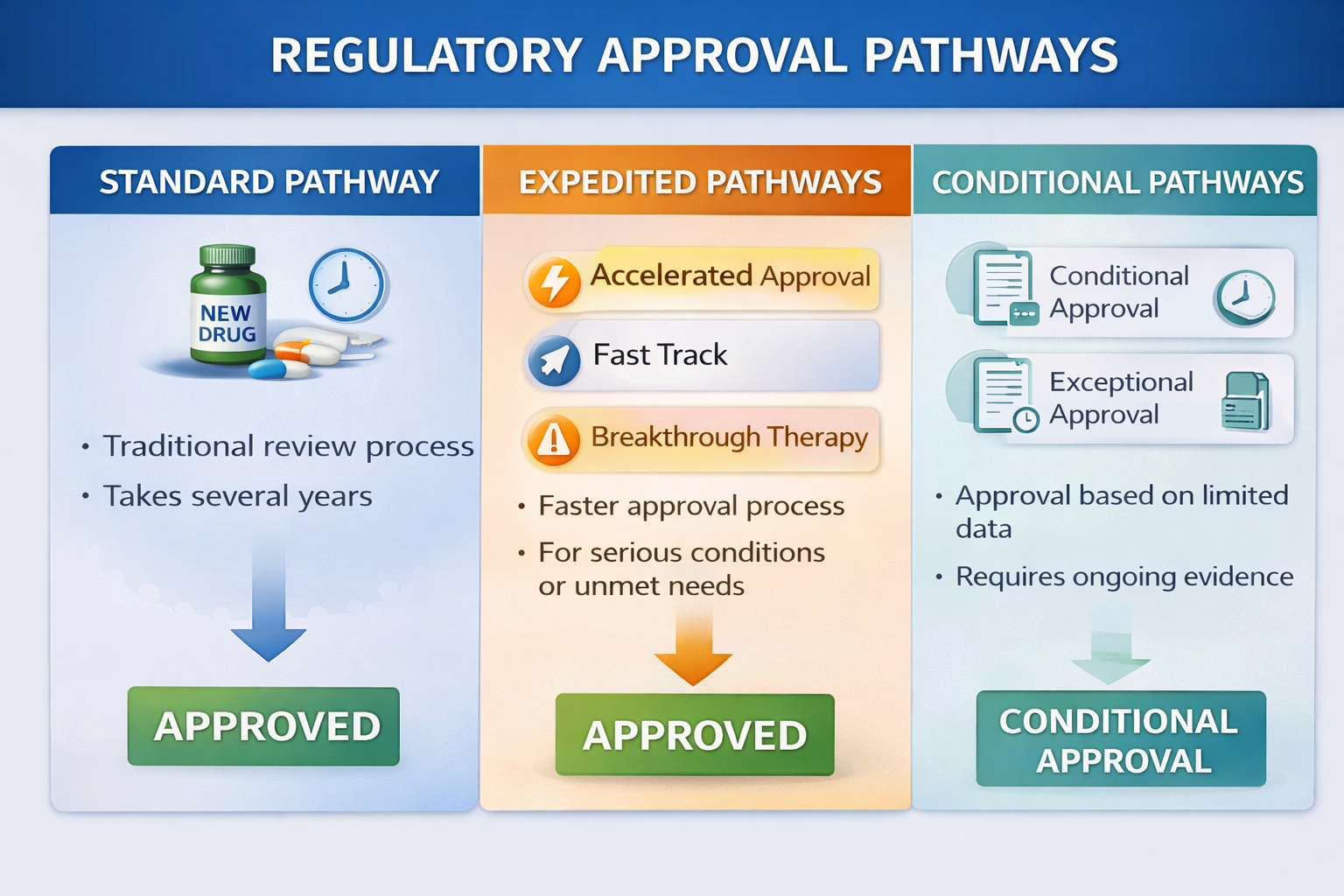 Standard (Full) Approval Pathway
Standard (Full) Approval Pathway
The standard approval pathway is the traditional route used for most new drugs and biologics. In this pathway, the sponsor must submit complete scientific data demonstrating safety, efficacy, and quality.
This includes Phase I, Phase II, and Phase III clinical trial data, full nonclinical (preclinical) data, comprehensive Chemistry, Manufacturing and Controls (CMC) information, and submission in CTD or eCTD format. Regulatory authorities conduct a complete benefit–risk assessment before granting marketing authorization.
This pathway is typically used for new chemical entities, new biologics, and standard therapeutic products that do not qualify for expedited programs.
Accelerated or Expedited Pathways
Accelerated pathways are designed for medicines that treat serious or life-threatening conditions or address unmet medical needs. These programs aim to shorten development and review timelines to provide faster patient access.
Key features may include shortened regulatory review timelines, early and frequent interaction with regulatory authorities, and rolling submissions where data is submitted in stages rather than all at once.
Examples include Fast Track, Breakthrough Therapy, and Priority Review programs in the United States, Accelerated Assessment in the European Union, and Priority Review pathways in Canada, the United Kingdom, and Australia.
Conditional Approval Pathways
Conditional approval allows medicines to be authorized based on less complete clinical data when the potential benefits outweigh potential risks. This pathway is commonly used for serious conditions with high unmet medical need.
Approval is granted on the condition that the sponsor conducts confirmatory post-approval studies, implements enhanced pharmacovigilance measures, and complies with strict post-marketing obligations.
Examples include Accelerated Approval in the United States, Conditional Marketing Authorization in the European Union, and Provisional Approval in Australia.
Generic Drug Approval Pathway
The generic approval pathway is intended for products equivalent to an already approved reference medicine. Instead of repeating full clinical trials, the sponsor must demonstrate bioequivalence with the reference product along with adequate quality and stability data and compliance with manufacturing standards.
Examples include the Abbreviated New Drug Application (ANDA) in the United States, the Abbreviated New Drug Submission (ANDS) in Canada, and generic drug approvals through CDSCO in India.
This pathway allows cost-effective alternatives while maintaining safety and quality standards.
Biosimilar Approval Pathway
Biosimilars are biologic products that are highly similar to an approved reference biologic. Due to the complexity of biologics, approval requires a stepwise comparability approach including detailed analytical similarity studies, supporting nonclinical data, and limited clinical studies to confirm that there are no clinically meaningful differences in safety, purity, or potency compared to the reference product.
Orphan Drug Pathway
The orphan drug pathway supports the development of medicines for rare diseases affecting small patient populations. Regulators provide incentives such as market exclusivity for a defined period, fee reductions, scientific guidance, and regulatory support throughout development.
Orphan drug programs are offered by agencies including the FDA, EMA, and MHRA.
Reliance and Recognition Pathways
Reliance pathways allow regulatory authorities to use or recognize evaluations performed by another trusted regulatory agency. This approach reduces duplication of work and speeds up access to medicines.
Examples include the International Recognition Procedure used by MHRA, the Comparable Overseas Regulator pathway used by TGA, and various reliance models used in emerging markets.
Key Evaluation Components Across All Pathways
Regardless of the specific approval route, regulatory authorities consistently evaluate core components including product quality (chemistry, manufacturing, and controls), nonclinical safety data, clinical evidence of efficacy and safety, and an overall benefit–risk assessment.
Authorities also review labeling and prescribing information to ensure accurate communication to healthcare professionals and patients. In addition, all approved products must comply with post-marketing risk management and pharmacovigilance requirements to monitor safety after market entry.
Regulatory approval pathways provide structured yet flexible mechanisms to evaluate medicines while maintaining strict standards of safety, efficacy, and quality, and supporting innovation and timely patient access to important therapies.
