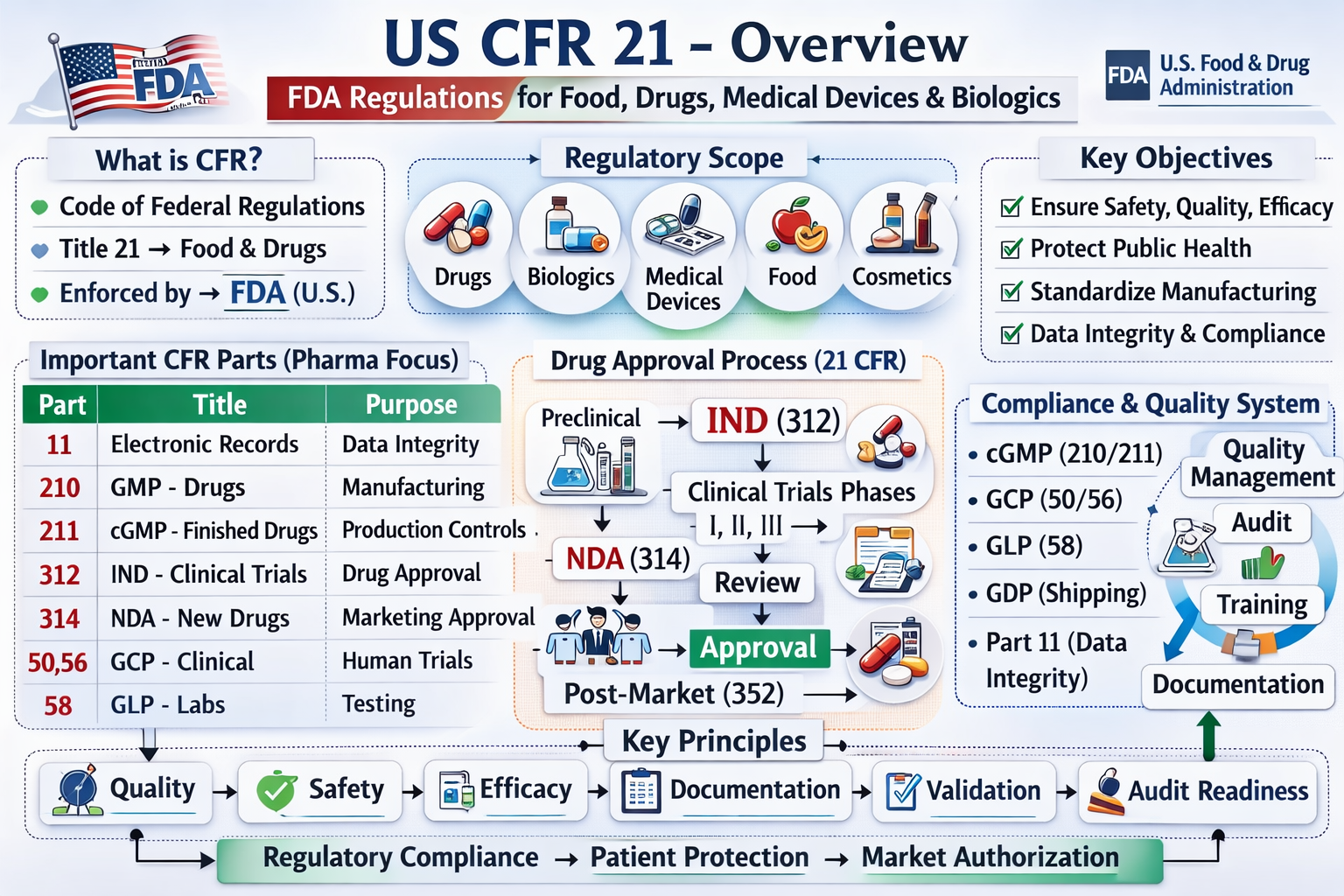
US CFR 21 Overview
Title 21 of the Code of Federal Regulations (CFR) – Legal and Operational Framework for Pharmaceutical Compliance
Title 21 of the Code of Federal Regulations (CFR) constitutes the primary set of rules governing food, drugs, biologics, medical devices, cosmetics, and tobacco products in the United States. These regulations are enforced by the U.S. Food and Drug Administration (FDA) and form the legal foundation for protecting public health. They define how regulated products must be developed, tested, manufactured, labeled, distributed, and monitored after approval. For professionals in pharmaceutical manufacturing, clinical research, quality assurance, and regulatory affairs, Title 21 is not merely a legal reference but a practical framework guiding daily operations. Understanding these regulations is essential to ensure that products are safe, effective, and compliant with federal standards.
The CFR is organized hierarchically, which facilitates navigation and practical application. At the top level, the Title represents a broad subject area, with Title 21 specifically covering FDA-regulated products. Within each title, Chapters indicate the regulatory authority, followed by Subchapters, which group related product categories or regulatory topics. Parts within each subchapter address specific regulatory requirements. For instance, Title 21 encompasses Food and Drugs. Chapter I represents the FDA. Subchapter C focuses on drugs, and Part 211 describes the Current Good Manufacturing Practice (CGMP) requirements for finished pharmaceuticals.
| Hierarchy Level | Example in Title 21 | Description |
|---|---|---|
| Title | 21 | Covers all FDA-regulated products including drugs, biologics, and medical devices |
| Chapter | I | Represents the FDA as the enforcing agency |
| Subchapter | C | Focuses on drugs |
| Part | 211 | Defines CGMP requirements for manufacturing finished pharmaceuticals |
Title 21 regulations cover the entire lifecycle of regulated products, forming a continuous chain of compliance from preclinical research through clinical trials, product approval, commercial manufacturing, and post-market surveillance. Preclinical studies must follow Good Laboratory Practice (GLP) to produce reliable data, supporting Investigational New Drug (IND) applications. Clinical trials are conducted under Good Clinical Practice (GCP), and approved products enter commercial manufacturing under Current Good Manufacturing Practice (GMP). Distribution and post-marketing activities are governed by additional GxP requirements to maintain quality, safety, and efficacy throughout the lifecycle.
Certain parts of Title 21 are particularly essential for pharmaceutical professionals. Part 11 addresses electronic records and electronic signatures, requiring system validation, secure access controls, and audit trails to ensure data integrity. Parts 210 and 211 define CGMP for drugs, establishing standards for personnel qualifications, facility design, equipment maintenance, production controls, and laboratory testing. Compliance ensures that each batch meets intended identity, strength, quality, and purity. Part 312 governs IND applications, including protocol submissions, safety reporting, and regulatory oversight during clinical trials. Part 314 describes New Drug Application (NDA) procedures, including labeling, post-approval changes, and regulatory communications. Parts 50 and 56 protect human subjects, specifying informed consent requirements and Institutional Review Board responsibilities.
| Part | Focus Area | Key Requirements |
|---|---|---|
| 11 | Electronic records and signatures | System validation, secure access, audit trails |
| 210 | CGMP general provisions | Facility design, personnel qualifications, production controls |
| 211 | CGMP for finished pharmaceuticals | Equipment maintenance, batch record integrity, laboratory testing |
| 312 | IND applications | Study protocols, safety reporting, regulatory oversight |
| 314 | NDA procedures | Marketing approval, labeling, post-approval submissions |
| 50 | Informed consent | Participant understanding of risks and benefits |
| 56 | Institutional Review Boards | Ethical oversight and approval of clinical trials |
The regulatory philosophy of Title 21 is built on four core pillars: quality, safety, documentation, and data integrity. Quality requires that systems ensure products consistently meet predefined standards through validated processes, controls, and audits. Safety mandates evaluation of every deviation, complaint, or adverse event for potential patient impact. Documentation ensures that every activity is recorded; if it is not documented, it is considered not to have occurred. Data integrity requires that all information is reliable, consistent, and traceable, achieved through controlled access, proper recordkeeping, and permanent audit trails.
FDA enforcement of Title 21 is carried out through routine and for-cause inspections of regulated facilities. Investigators assess compliance with applicable regulations and issue Form 483 for observed deficiencies. Serious non-compliance may lead to warning letters, import alerts, product recalls, or facility shutdowns. In industry practice, compliance is maintained through structured operational systems, including Corrective and Preventive Action (CAPA) programs, change management, document control, personnel training, and validation master plans. Audit readiness is a continuous requirement, not a one-time effort, ensuring that processes, documents, and personnel can withstand regulatory scrutiny at any time. Professionals must understand how regulatory requirements, such as Part 211, impact daily workflows, including batch record review, equipment validation, and laboratory testing procedures.
Title 21 CFR regulations provide a comprehensive legal and operational foundation for life sciences regulation in the United States. They govern every stage of the product lifecycle, from research and clinical trials to manufacturing and post-market monitoring. For regulatory affairs, quality, and clinical professionals, mastering Title 21 ensures compliance, protects patient safety, and supports the development, approval, and distribution of safe and effective medical products. Knowledge of Title 21 is fundamental for performing regulatory submissions, preparing for inspections, maintaining quality systems, and sustaining ongoing compliance in the pharmaceutical industry.
