NDA Types
NDA & Its Type
A New Drug Application (NDA) is the formal submission through which a sponsor seeks marketing approval for a drug product in the United States under Section 505 of the Federal Food, Drug, and Cosmetic Act. From a Global Drug Regulatory Affairs perspective, understanding NDA types is critical because each pathway has distinct data requirements, exclusivity implications, development timelines, and strategic considerations. For a job-ready regulatory professional, the ability to classify an NDA correctly determines submission strategy, intellectual property positioning, and lifecycle management planning.
The FDA recognizes multiple NDA types under Section 505(b) and 505(j). The major categories are summarized below.
Overview of NDA Classification
| NDA Type | Legal Basis | Data Requirement | Typical Applicant | Strategic Use |
|---|---|---|---|---|
| 505(b)(1) NDA | Section 505(b)(1) | Full reports of safety and efficacy | Innovator company | New chemical entity or full development program |
| 505(b)(2) NDA | Section 505(b)(2) | Partial reliance on existing data | Innovator or reformulation sponsor | Modified drug or new indication |
| 505(j) ANDA | Section 505(j) | Bioequivalence only | Generic manufacturer | Generic version of approved drug |
Each pathway reflects a different regulatory and commercial strategy.
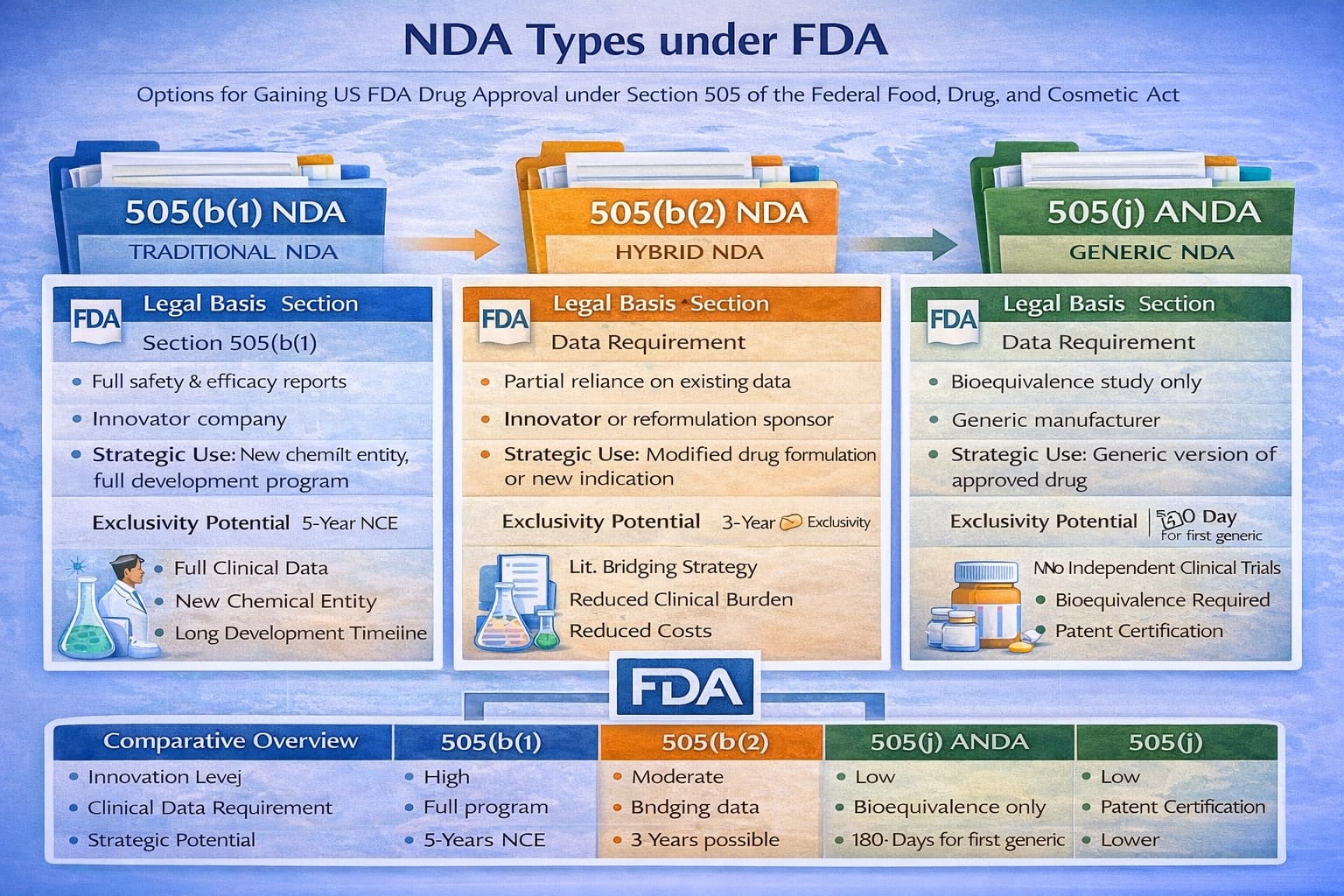
505(b)(1) NDA – Full NDA (Traditional NDA)
A 505(b)(1) NDA is the standard pathway for innovative drugs. The sponsor submits complete reports of investigations of safety and effectiveness conducted by or for the applicant. This includes full nonclinical, clinical, and CMC data generated during development under an active IND.
| Key Feature | Regulatory Implication |
|---|---|
| Full Clinical Development Program | Phase I–III trials required |
| Independent Data Ownership | No reliance on other sponsors’ data |
| Eligible for NCE Exclusivity | 5 years for new chemical entity |
| High Development Cost | Long timelines and significant investment |
This pathway is typically used for new molecular entities, biologically novel products (non-biologic small molecules), or drugs with entirely new therapeutic mechanisms.
505(b)(2) NDA – Hybrid NDA
The 505(b)(2) pathway allows the sponsor to rely in part on existing published literature or the FDA’s prior findings of safety and effectiveness for an approved drug. It bridges innovation and generic development.
| Key Feature | Regulatory Implication |
|---|---|
| Partial Data Reliance | Reduced clinical burden |
| Bridging Studies Required | PK or limited clinical trials |
| May Receive Exclusivity | 3 years for new clinical investigations |
| Strategic Reformulation Pathway | New dosage form, route, strength, or indication |
Common examples include extended-release formulations, new combinations, new routes of administration, or repurposed drugs for new indications. Regulatory professionals must carefully justify scientific bridging to the referenced listed drug.
505(j) ANDA – Abbreviated New Drug Application
The 505(j) pathway applies to generic drugs. Sponsors must demonstrate that their product is bioequivalent to a previously approved reference listed drug.
| Key Feature | Regulatory Implication |
|---|---|
| No Independent Clinical Efficacy Trials | Bioequivalence data sufficient |
| Pharmaceutical Equivalence Required | Same active ingredient, strength, dosage form |
| Patent Certification Required | Paragraph I–IV certification |
| 180-Day Exclusivity Possible | For first generic challenger |
Generic sponsors do not submit full clinical safety and efficacy data but must ensure strict equivalence in quality and performance.
NDA Sub-Classification by Review Type
In addition to legal classification, NDAs are categorized based on FDA review priority.
| Review Type | Timeline | Eligibility Criteria |
|---|---|---|
| Standard Review | 10 months | Typical new drug submission |
| Priority Review | 6 months | Significant improvement over existing therapy |
Priority review status is granted if the drug demonstrates meaningful advancement in safety or effectiveness.
Strategic Considerations for Regulatory Professionals
Selecting the correct NDA pathway requires careful regulatory intelligence analysis. Factors influencing decision-making include intellectual property status, available literature, exclusivity strategy, development budget, and competitive landscape.
For example, a reformulated product with improved compliance but same active ingredient may qualify under 505(b)(2), reducing development cost and timeline compared to a full 505(b)(1) program. Conversely, a completely new molecular entity requires the full traditional pathway.
Comparative Summary
| Parameter | 505(b)(1) | 505(b)(2) | 505(j) |
|---|---|---|---|
| Innovation Level | High | Moderate | Low |
| Clinical Data Requirement | Full program | Partial bridging | Bioequivalence only |
| Development Time | Long | Moderate | Short |
| Exclusivity Potential | 5 years (NCE) | 3 years possible | 180 days (first generic) |
| Cost | Very high | Moderate | Lower |
Job-Ready Competencies
A Regulatory Affairs Associate must understand the legal distinctions among NDA types, interpret FDA guidance, evaluate data bridging requirements, and support strategic submission planning. Familiarity with patent certifications, exclusivity periods, and lifecycle management planning is critical in pharmaceutical regulatory roles.
Conclusion
NDA classification determines the scientific, regulatory, and commercial trajectory of a drug product in the United States. Whether pursuing a full 505(b)(1) NDA, a hybrid 505(b)(2) pathway, or a generic 505(j) application, regulatory professionals must align development strategy with legal framework and business objectives. Mastery of NDA types is a foundational competency for careers in Drug Regulatory Affairs and global pharmaceutical development.
