IND Overview
What Is IND?
Investigational New Drug Application (IND) is the formal regulatory mechanism through which a sponsor seeks authorization from the US Food and Drug Administration to initiate clinical trials of a new drug in the United States. From a Global Drug Regulatory Affairs perspective, the IND is not merely a submission document; it is a strategic regulatory milestone that bridges preclinical development and human clinical research. For a beginner aiming to become job-ready, it is essential to understand that the IND is both a scientific justification and a regulatory risk management document designed to demonstrate that a drug is reasonably safe for initial testing in humans.
The legal basis for the IND framework is derived from the Federal Food, Drug, and Cosmetic Act and codified under 21 CFR Part 312. Before any clinical investigation begins in the United States, an IND must be in effect unless the study qualifies for exemption. An IND becomes effective 30 calendar days after the FDA receives the submission, unless the agency places the study on clinical hold. Therefore, regulatory professionals must ensure that the submission is scientifically robust, complete, and strategically aligned to avoid delays.
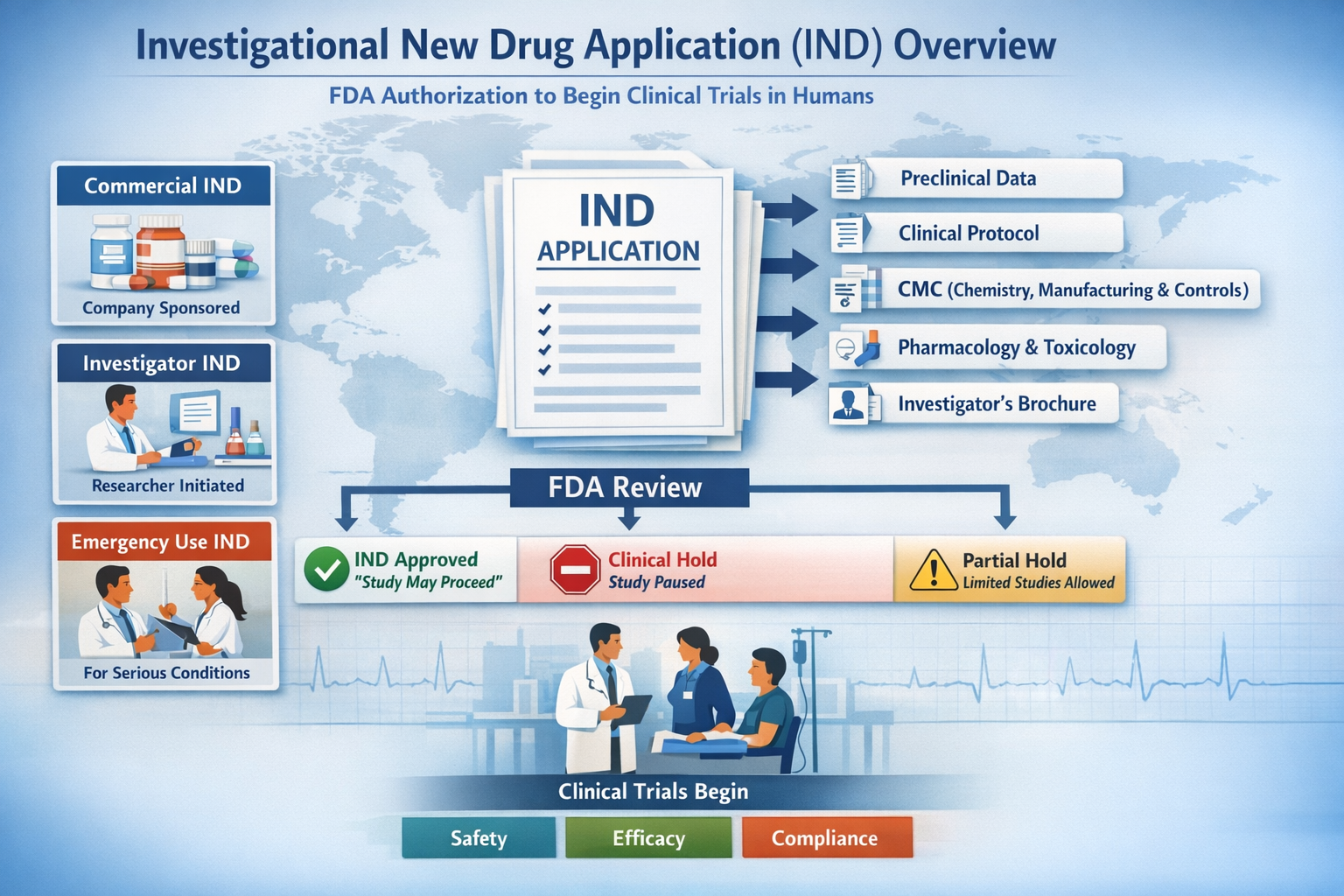
There are three primary types of IND applications, each serving a distinct regulatory purpose.
| IND Type | Sponsor | Purpose | Typical Industry Use |
|---|---|---|---|
| Commercial IND | Pharmaceutical/Biotech Company | To support marketing approval | Standard pathway for innovative drugs |
| Investigator IND | Individual Investigator | Investigator-initiated studies | Academic or hospital-based trials |
| Emergency Use IND | Physician | Emergency treatment for serious conditions | Single patient expanded access |
A job-ready regulatory professional must clearly understand the structure and content of an IND submission. While the Common Technical Document format is used globally for marketing applications, the IND follows a specific U.S. structure aligned with CTD principles but organized differently. The core components are summarized below.
| Section | Content Overview | Regulatory Objective |
|---|---|---|
| Introductory Statement | Drug description, rationale | Establish development intent |
| Investigator’s Brochure | Preclinical and clinical data summary | Inform investigators |
| Protocols | Study design, endpoints, safety monitoring | Ensure subject protection |
| Chemistry, Manufacturing and Controls (CMC) | Drug substance and product details | Assure product quality |
| Pharmacology and Toxicology | Animal safety data | Justify first-in-human exposure |
| Previous Human Experience | If applicable | Support risk evaluation |
| Additional Information | IRB details, financial disclosures | Ensure compliance |
The CMC section is often underestimated by beginners but represents a frequent cause of regulatory queries. The FDA expects sufficient information to ensure that the investigational product can be consistently manufactured with acceptable quality. This includes drug substance characterization, specifications, analytical methods, stability data, and manufacturing process description. Even at early development stages, a basic level of GMP compliance must be demonstrated.
The pharmacology and toxicology section is scientifically critical. Before first-in-human trials, sponsors must conduct GLP-compliant toxicology studies in at least two species, typically one rodent and one non-rodent. Key toxicology endpoints include single-dose toxicity, repeat-dose toxicity, genotoxicity, and safety pharmacology. Dose selection for Phase I trials is based on the No Observed Adverse Effect Level (NOAEL) and calculation of the Human Equivalent Dose (HED). Regulatory professionals must collaborate closely with nonclinical scientists to justify safe starting doses and dose escalation strategies.
The clinical protocol is the heart of the IND. It must define study objectives, inclusion and exclusion criteria, dosing regimen, safety monitoring plan, stopping rules, and statistical considerations. Regulatory professionals must ensure alignment between nonclinical findings and clinical design. For example, if toxicology studies reveal potential hepatotoxicity, liver function monitoring must be incorporated into the protocol.
The IND review process is time-bound and risk-based. Within 30 days, the FDA evaluates safety, scientific rationale, manufacturing controls, and subject protection measures. The possible outcomes are summarized below.
| FDA Decision | Meaning | Regulatory Action Required |
|---|---|---|
| IND Effective | Study may proceed | Begin clinical trial |
| Clinical Hold | Safety or quality concerns identified | Address deficiencies and resubmit |
| Partial Hold | Certain protocols restricted | Amend specific study components |
Clinical holds can arise from insufficient toxicology data, inadequate CMC controls, unreasonable clinical design, or investigator qualification concerns. Therefore, proactive pre-IND meetings with the FDA are strategically recommended. A pre-IND meeting allows sponsors to obtain regulatory feedback on study design, nonclinical plans, and CMC development before formal submission.
Post-IND responsibilities are equally important. Once the IND is active, sponsors must maintain regulatory compliance through amendments and periodic reporting.
| Submission Type | Purpose | Timeline |
|---|---|---|
| Protocol Amendment | Add or modify study protocols | Before implementation |
| Information Amendment | Submit additional data | As needed |
| IND Safety Report | Report serious adverse events | Within 7–15 days |
| Annual Report | Summarize yearly progress | Within 60 days of anniversary |
From a global perspective, professionals must recognize that while the IND is specific to the United States, similar frameworks exist in other jurisdictions. In Europe, Clinical Trial Applications are submitted to national authorities under EMA coordination, while in India, permissions are obtained from CDSCO. However, the scientific foundation remains consistent: demonstration of safety, quality, and ethical study conduct.
For career readiness, an entry-level Regulatory Affairs Associate working on INDs must develop competencies in regulatory writing, scientific data interpretation, eCTD publishing basics, cross-functional coordination, and regulatory intelligence tracking. Familiarity with FDA guidance documents, ICH guidelines such as ICH M3(R2) for nonclinical safety studies, and GCP principles is mandatory. In leading companies such as Pfizer, Novartis, Roche, and IQVIA, IND preparation is a collaborative, matrix-driven process involving regulatory, clinical, nonclinical, CMC, quality assurance, and biostatistics teams.
In conclusion, the Investigational New Drug Application is the gateway to human clinical research in the United States. It is not simply a regulatory formality but a scientifically rigorous, strategically structured submission that defines the success trajectory of drug development. Mastery of IND preparation and lifecycle management is a core competency for any aspiring Drug Regulatory Affairs professional aiming to work in global pharmaceutical organizations.
