CTD Overview and Purpose
Common Technical Document (CTD – Overview and Purpose)
Introduction
The Common Technical Document, or CTD, is the globally accepted format for regulatory submissions of pharmaceuticals, biologics, and biosimilars. Developed under the International Council for Harmonisation (ICH), the CTD was introduced to harmonize the submission of quality, safety, and efficacy data across regulatory authorities such as the US FDA, EMA, Health Canada, TGA, and CDSCO. Prior to the CTD, each regulatory agency required a unique submission format, which resulted in redundancy, inefficiencies, delays, and increased risk of errors. The CTD allows a single dossier to be submitted to multiple regions with minimal modifications, ensuring faster and more consistent regulatory review. It is designed to support both innovator and generic products, facilitating lifecycle management, including post-approval variations, renewals, and safety updates. For Regulatory Affairs professionals, the CTD provides a structured framework to organize complex data in a manner that meets global regulatory standards, improves cross-functional collaboration, and enhances communication with agencies.
Purpose of the CTD
The CTD was created to standardize and streamline the preparation, submission, and review of regulatory dossiers. Its primary purpose is to provide a single, harmonized structure that presents all critical information about a medicinal product in a clear, consistent, and accessible format. By standardizing submissions, the CTD allows regulatory authorities to focus on scientific evaluation rather than interpreting varying submission formats, thus reducing review time. For industry professionals, the CTD reduces duplication of effort when preparing dossiers for multiple regions, supports electronic submission via the eCTD format, and enables modular updates to facilitate changes in manufacturing, labeling, or clinical data. The CTD also serves as a central reference for inspection readiness, regulatory queries, and internal audits. Mastery of the CTD ensures that professionals can plan, compile, and manage dossiers efficiently, making it a foundational skill for any job-ready Regulatory Affairs candidate.
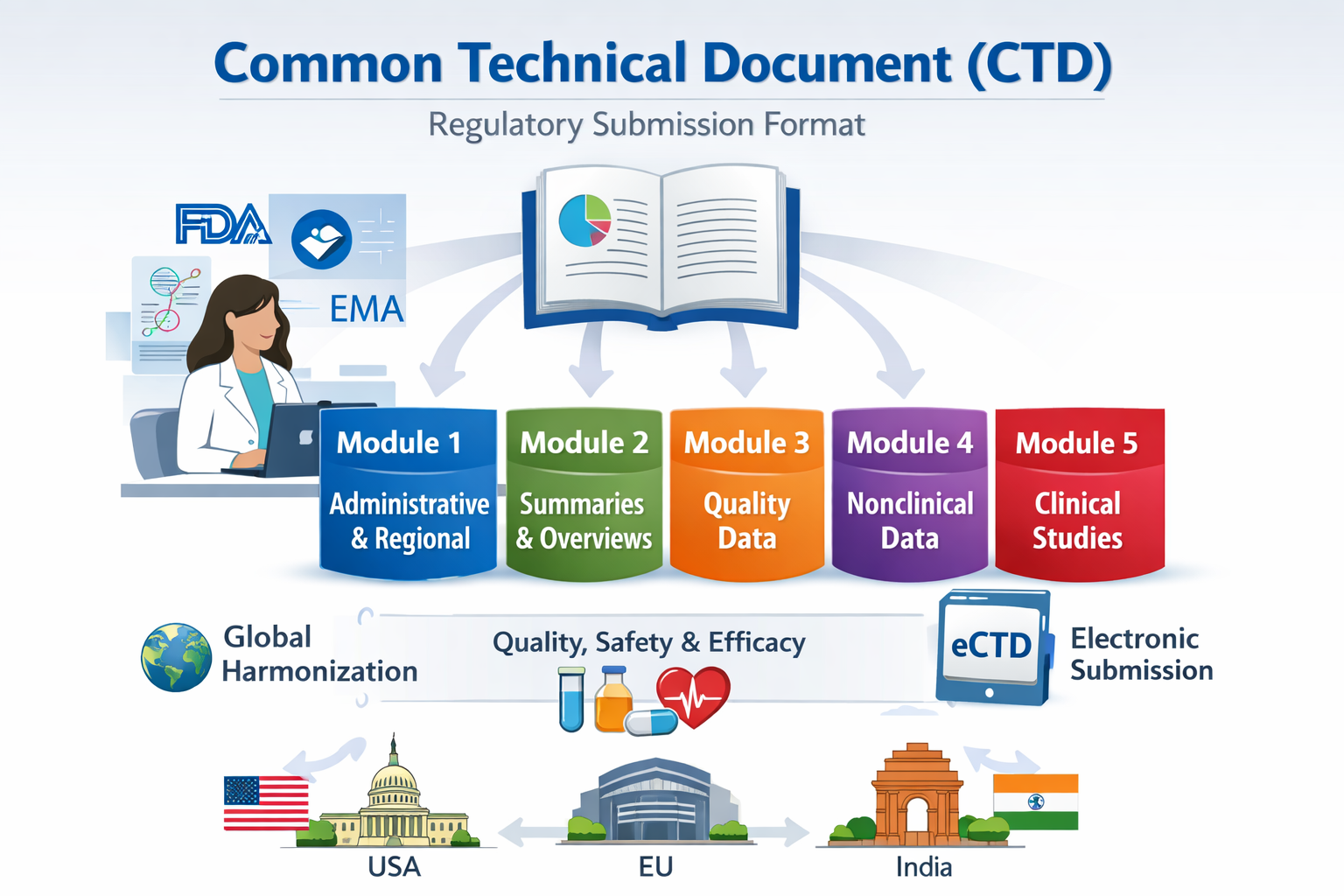
CTD Structure
The CTD is organized into five modules. Each module has a defined purpose and contains specific types of information that together provide a comprehensive view of a drug’s quality, safety, and efficacy. The modular structure improves clarity, facilitates submission across multiple jurisdictions, and supports both paper-based and electronic submissions. The five modules are summarized below:
| Module | Content Overview |
|---|---|
| Module 1 | Administrative and regional-specific information. Includes application forms, cover letters, labeling, prescribing information, and other region-specific documents. Module 1 is unique to each jurisdiction and is not harmonized. |
| Module 2 | Summaries and overviews of data presented in Modules 3, 4, and 5. Contains the Quality Overall Summary, Nonclinical Overview, Clinical Overview, and integrated summaries. Module 2 provides regulators with a high-level understanding of the data, facilitating review and decision-making. |
| Module 3 | Quality information covering Chemistry, Manufacturing, and Controls (CMC). Includes detailed information about the drug substance and drug product, manufacturing processes, analytical methods, specifications, stability studies, and packaging. This module demonstrates product quality, consistency, and compliance with GMP requirements. |
| Module 4 | Nonclinical study reports including pharmacology, pharmacokinetics, and toxicology. Data in this module support the safety evaluation of the investigational or marketed product. It demonstrates that the product has an acceptable safety profile before and during clinical trials. |
| Module 5 | Clinical study reports including Phase I to Phase IV trials. Contains efficacy and safety data, integrated summaries, statistical analyses, and a comprehensive risk-benefit assessment of the product. Module 5 is critical for demonstrating clinical performance and supporting regulatory approval. |
Integration of CTD in Regulatory Strategy
The CTD is more than a submission format; it forms the foundation of a regulatory strategy. Regulatory Affairs professionals use the CTD to plan the sequence of submissions, identify regulatory requirements for each region, and anticipate potential questions from authorities. It allows teams to coordinate data collection across quality, clinical, and nonclinical functions, ensuring that information is complete and internally consistent. In global submissions, the CTD enables companies to prepare a core dossier that can be adapted for specific regions, reducing preparation time, minimizing errors, and ensuring compliance with local regulations.
Practical Applications in Regulatory Affairs
Mastery of the CTD equips professionals to handle all stages of regulatory submissions. It allows them to prepare dossiers efficiently for new drug approvals, generic applications, and post-marketing changes. Professionals can manage cross-functional inputs, convert submissions to electronic formats (eCTD), and respond effectively to deficiency letters or regulatory queries. The CTD structure supports lifecycle management, including annual product renewals, labeling changes, and submission of post-marketing safety updates. In practice, understanding the CTD enables Regulatory Affairs professionals to plan timelines, assign responsibilities, and ensure that dossiers meet global standards, which is essential for job-ready candidates seeking roles in pharmaceutical companies, contract research organizations, and regulatory consulting firms.
Conclusion
The Common Technical Document is a cornerstone of modern regulatory submissions. It provides a globally harmonized framework that integrates quality, safety, and efficacy data in a clear, consistent, and standardized manner. Mastery of the CTD is critical for Regulatory Affairs professionals, as it enables efficient dossier preparation, cross-functional collaboration, and successful interactions with regulatory authorities. Understanding both the purpose and structure of the CTD prepares candidates to manage global submissions, address regulatory challenges, and contribute to the approval and lifecycle management of pharmaceutical products. A deep understanding of CTD principles ensures that job-ready professionals can confidently support regulatory strategies, maintain compliance across regions, and facilitate the successful approval of drugs worldwide.
